アンジェルマン症候群は小児慢性特定疾病に指定されている染色体異常で、出生前診断の一つであるNIPTを受けた際に発覚することがあります。
ここではアンジェルマン症候群について、どのような疾患なのか?その症状や原因などについて基本的な情報をご紹介いたします。
アンジェルマン症候群とは

アンジェルマン症候群とは、多くの遺伝情報が詰まった染色体の異常によって起こる先天性の疾患です。
重度の精神遅滞を中心とする中枢神経機能障害が主な症状です。
【アンジェルマン症候群の主な特徴】
- 重い知的障害
- 重い言語障害
- 運動障害
- てんかん
- ぎこちない動き(失調性歩行)
- ちょっとしたことで容易に笑う
- 多動性
- 特徴的な顔貌
基本的には命に直接かかわるような合併症はなく、誤嚥性肺炎や歩行時の転倒による事故などを除けば平均寿命は一般の人と変わりません。
染色体異常と聞くと遺伝するのでは?と思われるかもしれませんが、ほとんどの場合は遺伝ではなく遺伝子の突然変異によって偶然に起こり、だれにでもその可能性はあります。
原因
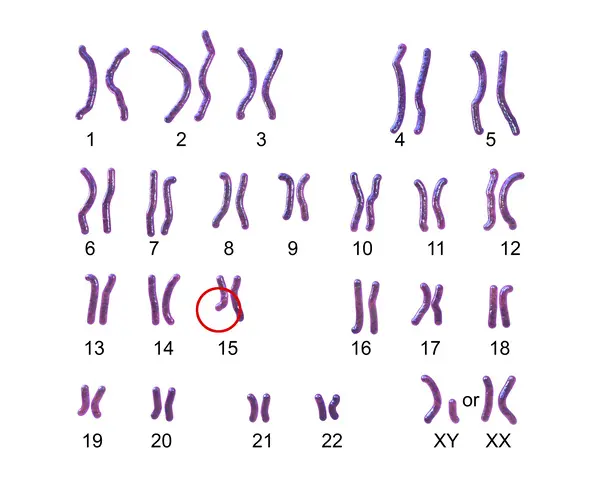
アンジェルマン症候群は染色体異常の一つです。
染色体とは、遺伝子を含むDNAが、折り重なってできたものです。
ヒトの体細胞には46本の染色体があります。
そのうちの44本はペアで22対の「常染色体」といい、
あとの2本は男女の性別を決める「性染色体」といいます。
染色体は父親と母親から1本ずつもらったものがペアとなっています。
常染色体は長いものから順番に1~22番の番号が割り振られています。
そのうちの15番染色体に存在する、「UBE3A」という遺伝子の働きが失われることでアンジェルマン症候群は発症します。
「UBE3A」遺伝子とは、父母から1本ずつ受け継いだ染色体のうち、母方から受け継いだものだけがはたらくという遺伝子です。
遺伝子は通常、父母から一本ずつ受け継がれたものがセットになっており、どちらの親由来のものでも同じようにはたらきます。
しかしいくつかの遺伝子については父親、もしくは母親から受け継いだ遺伝子のみがはたらくことが知られており、これを「ゲノム刷り込み現象」といいます。
アンジェルマン症候群は、母親由来の「UBE3A」遺伝子に異常が起こりはたらかなくなってしまうために起こります。
父親由来の「UBE3A」は「ゲノム刷り込み現象」によりもともとはたらくことができませんので、「UBE3A」の機能は失われてしまいます。
同じくゲノム刷り込み現象によって起こる疾患に「プラダーウィリー症候群」がありますが、こちらは父親由来の特定の遺伝子の機能喪失によって起こります。
アンジェルマン症候群は発症のメカニズムにより大きく分けて4つのタイプがあります。
基本的にアンジェルマン症候群は親から子へ遺伝しませんが、タイプによっては遺伝する可能性のあるものがあります。
なお、約10%はどのタイプとも分からず、原因不明です。
欠失型
母親由来の15番染色体が一部欠損することにより、UBE3Aの働きが失われることにより起こります。
アンジェルマン症候群の約70%がこのタイプです。
基本的には偶然起こるもので、遺伝ではありません。
欠失型が一番症状が重い傾向にあります。
UBE3Aの突然変異
母親から受け継いだ15番染色体に存在するUBE3Aに変異があるために起こります。
アンジェルマン症候群の約11%がこのタイプです。
遺伝する可能性があります。
父親由来のUBE3Aに突然変異がある場合は、受け継いだ本人は発症はしませんが保因者ということになり、さらにその子供へと遺伝する可能性があります。
刷り込み変異
母親から受け継いだ15番染色体の刷り込み中心と呼ばれる領域に異常がある場合に起こります。
そのために、母親由来のUBE3Aがあったとしても感知できませんので、UBE3Aははたらくことができません。
アンジェルマン症候群の約5%がこのタイプです。
遺伝する可能性があります。
片親性ダイソミー
通常は父と母から15番染色体を1本ずつ受け継ぐはずのところを、父親から2本受け継ぐことにより、母親由来のUBE3Aが存在せず起こります。
片親性(かたおやせい)ダイソミーとは、2本の染色体を片親のみから受け継いだ場合をいいます。
アンジェルマン症候群の場合は父親から2本もらった状態ですが、母親から2本もらった場合は「プラダ―・ウィリー症候群」と呼び、症状が異なります。
アンジェルマン症候群の約3%がこのタイプです。
これは偶然に起こるもので、遺伝ではありません。
症状は欠失と比べて軽い傾向にあります。
アンジェルマン症候群の症状と特徴

アンジェルマン症候群は身体的な合併症は少ないですが、重い知的障害や言語障害があります。
【すべてのアンジェルマン症候群の人に見られる症状】
知的障害(知的能力障害)
知的障害とは、日常生活で頭脳を使う知的行動に支障があることをいいます。
言語障害
まったくしゃべらないか、話せても「あー」などの短い言葉や単語に限られ、文章を話すことは出来ないため言葉によるコミュニケーションは難しいでしょう。
失調性歩行
ふらふらと不安定でぎこちない歩き方をいいます。
動作のバランスが悪く、ぎくしゃくとした歩き方をします。
容易に笑う
ちょっとしたことでもすぐに笑ったり、頻繁に声をたてて笑います。
特異な行動
手を羽ばたかせたり手をたたく動作をよくみせます。
歩くときに腕を上げたりします。
すぐに興奮する性格で、好奇心旺盛です。
よく動き落ち着きがなく、集中力に欠ける点があります。
【アンジェルマン症候群の80%の人に見られる症状】
てんかん
幼少期に起こりやすい症状で、手足が震えるなどけいれん発作などがみられます。
てんかんとは、けいれんや突然意識がなくなるなどの「てんかん発作」を繰り返す状態をいいます。
小頭症
生まれたころは正常ですが、成長とともに頭囲の発達の遅れがみられ、2歳頃までに基準値と比べて頭囲が小さめ、もしくは小さいことがわかります。
【アンジェルマン症候群の20~80%の人に見られる症状】
睡眠障害
眠れなかったり早い時間に目が覚めたり、夜中に目が覚めやすかったりします。
睡眠の必要性が低下します。
弄舌癖(ろうぜつへき)
舌を突き出したり吸ったりする癖があります。
舌の使い方が下手で、飲み込むのが苦手だったりします。
また、過度に噛んだりもぐもぐする行動がみられます。
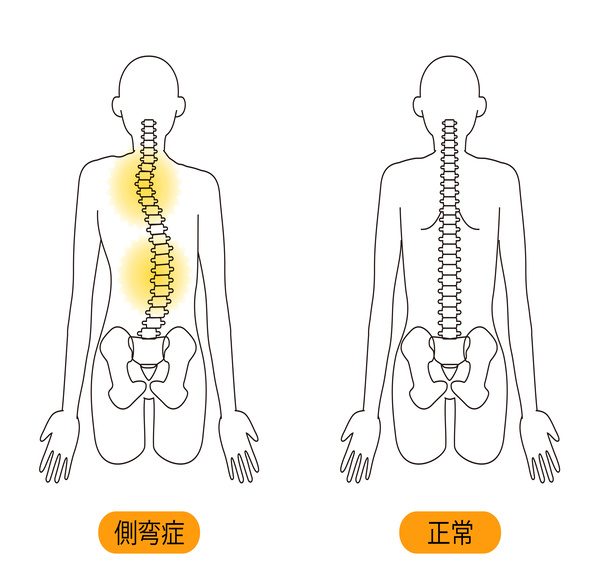
側弯症(そくわんしょう)
背骨が左右に曲がっている状態です。
子供のころにみられやすく、成人期にも少し進行する可能性があります。
そのほかにこのような特徴がみられます。
- 頻繁によだれを垂らす
- がにまたで歩く
- 斜視
- 色素が薄い(肌や髪、目など)
- 水に対する強い興味を示す
見た目の特徴

アンジェルマン症候群は、特徴的な顔貌があることが知られています。
- 顎が突き出てとがっている
- 口が幅広く大きい
- 歯と歯の隙間があいている
- 後頭部が扁平である
- 舌が突き出ている
もちろん個人差がありますので、その特徴が顕著なこともあれば、ほとんどわからない場合もあります。
発達・予後
アンジェルマン症候群の赤ちゃんは生まれてから半年~1年くらいで発達の遅れが目立ち始めます。
乳児期には、哺乳がうまくできず体重が増えづらいことがあります。
自分の頭を支えられない、立ち上がれないなどの運動の遅れがみられます。
だいたい5歳くらいで一人歩きが出来るようになりますが、不安定でバランスが悪く、ぎくしゃくした動きをします。
哺乳や嚥下の問題があると、胃食道逆流症などの胃腸の問題が伴いやすくなります。
言葉の遅れがあり、文章で話すことはできませんが、理解は進みます。
非言語によるコミュニケーションの訓練が大切です。
よく笑うことが特徴の一つで、ちょっとしたことで頻繁に声を立てて笑ったり、場にそぐわない愉快なふるまいをしたりします。
好奇心旺盛で、特に水やビニールなどのきらきらしたものに対する興味が強いため、思わぬ事故につながる恐れがあります。
注意欠如・多動症が見られることもあり、集中力や注意が続かない、落ち着きがないといった特徴があります。
自分の手やおもちゃなど、何でも口に入れてしまいます。
多くの人は歩けるようになりますが、膝の曲げ伸ばしが上手くできなかったり、足の裏が外側を向いている外反足(がいはんそく)であったりするため、スムーズには歩けません。
約10%の人は歩けないままだと言われています。
可動性の問題などで大人になるとあまり動かなくなることが多く、肥満になりやすいため注意が必要です。
便秘や胃食道逆流症はよく見られます。
アンジェルマン症候群の人は発達は遅れますが、早期療育により改善していく問題もあります。
アンジェルマン症候群の子が生まれる確率
生まれてくる赤ちゃんのうち、15,000~20,000人に1人の確率でアンジェルマン症候群であるといわれています。
日本では1,000人~3,000人ほどのアンジェルマン症候群の方が生活されているようです。
寿命
アンジェルマン症候群の平均寿命は一般の人と同じです。
染色体異常の中でも、新型出生前診断で検査をする「18トリソミー」や「13トリソミー」は生命予後が不良で、生まれてすぐに亡くなる赤ちゃんが多いのですが、アンジェルマン症候群は命に直接関わるような心臓の奇形などの合併症があるわけではありません。
ただし、歩行時の転倒や水に対する強い興味からくる水難事故などの可能性があり、注意が必要です。
遺伝するのか?

染色体異常と聞くと遺伝するのではと思われるかもしれませんが、ほとんどの場合は突然変異で、遺伝ではありません。
これは誰にでも起こる可能性があり、妊娠前の生活習慣や妊娠時の行動が原因になるわけではありません。
しかし稀に親から子へ遺伝する例もあり、アンジェルマン症候群では、「UBE3A突然変異」「刷り込み変異」の2タイプが遺伝する可能性があります。
検査・診断
臨床的な特徴がありアンジェルマン症候群の疑いがある場合は染色体検査を行ないます。
しかし、アンジェルマン症候群の典型的な症状があっても遺伝子検査で診断が出来ず原因不明なケースが約10%程度あります。
出生前診断
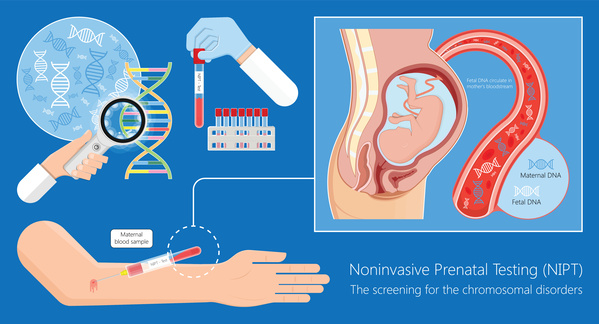
遺伝する可能性が事前に分かっている場合は出生前診断で検査を行いますが、そうでなければダウン症など他の染色体異常を調べるために受けた新型出生前診断(NIPT)や、羊水検査などで偶然発覚する場合もあります。
出生後の検査
アンジェルマン症候群の赤ちゃんは、見た目で分かるような合併症はほとんどなく、1歳頃になり特徴的な症状が現れてその疑いが持たれます。
精神や言葉の遅れがあってもアンジェルマン症候群だと分からず、診断が下るまで数年かかることもあります。
出生前の検査でアンジェルマン症候群だと分かったら?
出生前診断でダウン症などの染色体異常が発覚した場合、中絶を選択する人が多いことについて議論がつきません。
出生前診断の本来の目的は、妊娠中にお腹の赤ちゃんの病気や障害を見つけ、安全に分娩できる環境を整えて生まれた後の治療や生活環境の準備につなげるためのものです。
出生前の検査でアンジェルマン症候群だと分かったら。。。?
おそらく気が動転してしまい、不安を抱くことと思います。
まずは専門家による遺伝カウンセリングを受けて、状況を整理し正しい情報の元であなたとあなたのパートナーとでしっかり話し合うことが大切です。
治療・療育

アンジェルマン症候群の原因である染色体異常そのものに対する根本的な治療法はありません。
症状に合わせて対症療法を行なっていきます。
てんかん発作に対しては抗てんかん薬を使用することで、発作のない生活ができることが一般的です。
睡眠障害がひどい場合は、睡眠薬を使用します。
言葉の遅れに対しては言語療法を、運動機能や脳機能の遅れに対しては理学療法や作業療法を行ないます。
言語療法では、文章を話せるようになることは難しいため、身振りや道具を用いた非言語的なコミュニケーション法を身に付けます。
運動不足と食欲過剰による肥満が起こりやすく本人ではコントロールが難しいため、サポートが必要です。
早い段階から療育を行うことで、出来ることの幅が広がる可能性があります。
意思疎通を図れたり歩いたりできれば、学校に通うことも可能になります。
歩けない場合でも、肢体不自由特別支援学校というものがあります。
一般的な職場への就労は難しいかもしれませんが、障害者雇用での就労が考えられます。
まとめ

アンジェルマン症候群は染色体異常によって起こる先天性の病気です。
まだ解明されていないことも多く、現代医学では根本的な治療法はありません。
ご家族のサポートはもちろん、学校や自治体のサポートが不可欠です。
治療やケアは早い段階から始めることで、生活の質の向上が期待されます。
生まれる前にアンジェルマン症候群だと分かると、どんな疾患か分からず不安になるでしょうが、アンジェルマン症候群の家族会などもありますので、まずは一人で抱え込まずに遺伝カウンセラーや、家族会の方に相談してみるのがよいでしょう。
NIPT JapanのNIPTでは胎児のアンジェルマン症候群の可能性について調べることができます。